Mascot Distiller de novoでパズルを解く
EUPA(EUropean Proteomics Association)とLBMSDG(London周辺のProteomics研究者たちの勉強会グループ、LPDG内にあるDiscussion groupの1つ、London Biological Mass Spectrometry Discussion Group)が主催するMS/MSデータ解析チャレンジをご存知でしょうか?もしあなたがスペクトルからペプチド配列を直接読み取れるほどのエキスパートでしたら、是非ここで読むのをやめてください!
データ解釈の力や暗算能力が少し錆びついている大半の方には、Mascot Distillerと少しの推測力を使ってMS/MSデータ解析チャレンジの2つ目の課題の解決策を検討する過程を詳細に説明する事がお役に立つかもしれないと考え、今回ここでご紹介したいと思います。まだDistillerをお持ちでない方は、30日間の無料トライアルをご利用頂けます。(日本でのユーザーの方はマトリックスサイエンス社日本法人までお問い合わせください。)
課題となっているデータは、ラベル付きスペクトルが表示されたPDFファイルとして提示されています。このPDFデータからピークリストに変換する作業が、練習問題の最も面倒な作業です。PDFの表示方法によっては、CTRL+AとCTRL+Cを入力してすべてのテキストを選択とクリップボードへのコピーができます。これでうまくいったら、テキストをスプレッドシートに貼り付けて、軸のラベルやタイトルに当たる情報を削除してきれいにします。この方法がうまくできない場合はあきらめて手作業で1つずつ質量の値を入力すればいいだけです。
また、各行の強度値も必要です。Distillerでのde novo sequencing計算は強度の値を気にしないので、2列目の数字は全部同じ値を適用してもOKです。それでは気が済まない方は、あなたが思うピークの高さの推定値を入力してください。ただし値が正確である必要はありません。また、質量の数値を元にソートした方がきっと気持ちが良くて良いかと思いますが、Distillerにとっては必須ではありません。データをタブ区切りテキストとして保存します。お急ぎの場合は、こちらで準備したピークリストファイルをダウンロードしてご利用ください。
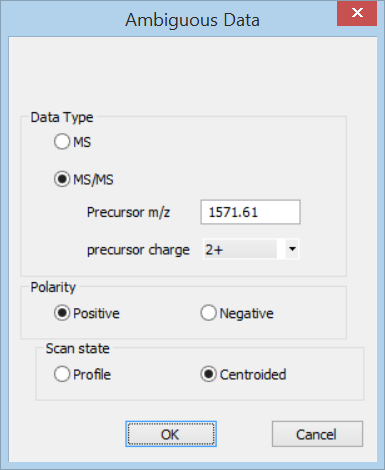
Distillerでは、menuのFile→New Project→Textを選択します。ここで先ほど作成したピークリストファイルを選択し、続いて現れるAmbiguous dataダイアログで下図のようにprecursorの情報を入力します。
これでDistillerにスペクトルが表示され、ピークピッキングの準備が整いました。しかし計算を進める前にいくつかの微調整が必要です。設定変更の画面である、Processing Optionsダイアログを開きます(menuのProcessing→Processing Options)。MS/MS processingタブで、”Useprecursor charge as maximum”のチェックを外して、すぐその上の設定項目”Maximum charge”を1に設定します。続いてMS peak pickingタブの右上”General”で、 ”Apply baseline correction”のチェックが入っていればこれを外し、すぐ下の”Fit method”を“Single peak”に変更します。また左下の”Peak profile”の下にある3つの幅の値をすべて0.02に設定します。
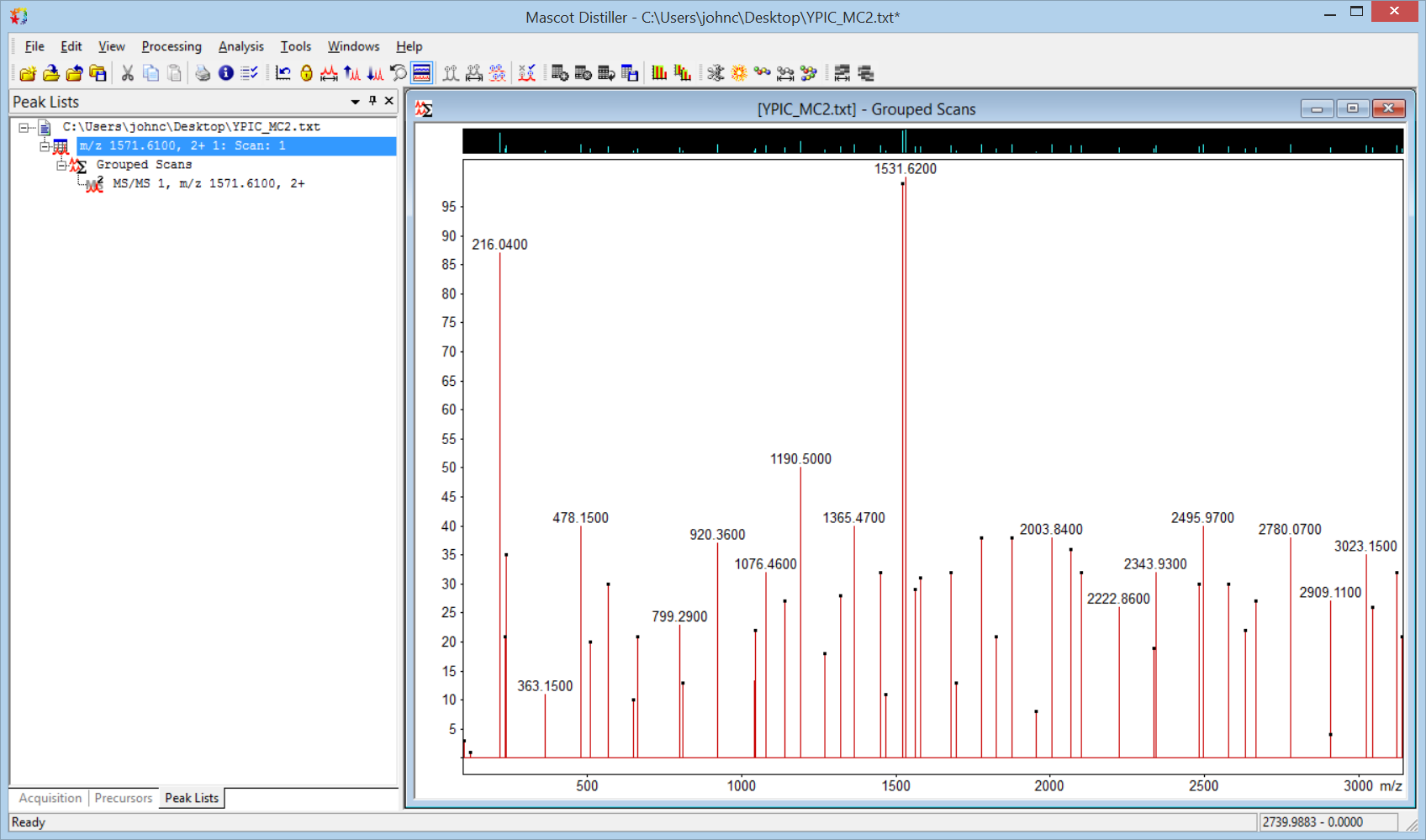
設定が終わったらOKボタンを押してダイアログを閉じ、ProcessingメニューまたはツールバーからProcess scanを選択します。これで以下の図のような結果が得られるはずです。
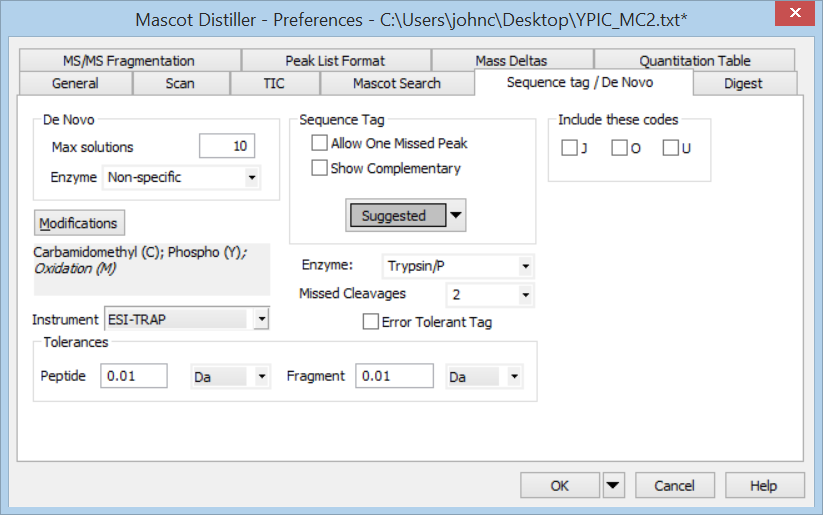
メニューのTools→Preferencesでダイアログを開き、Sequence tag / De Novoタブを選択します。今回の解析データがトリプシンペプチドであると断言できないので、”Enzyme”を”Specific and non-specific”に設定します。ペプチドは未知の方法で修飾されていると予め注意書きがあり、現段階 でprecursorの質量でもって解に縛りを与えたくないため、Peptide toleranceを5 Daに変更します。 一方Fragment toleranceについては0.1Daに変更しました。これは、すべてのラベルが小数点以下2桁で表示されている事から考えた推測です。OKボタンを選択しダイアログを閉じて、AnalysisメニューからDenovo searchを選択します。
計算すると、いくつかの類似した候補の解答が表示されます。うち上位3つは以下の通りです。
PHqDS[YA|SF|PH|MC][WG|SR|NE|Dq]PEPTPMEENR[EE]PSDE[SK]
PHqDS[YA|SF|PH|MC][WG|SR|NE|Dq]PEPTiDEENR[EE]PSDE[SK]
PHqDS[YA|SF|PH|MC][WG|SR|NE|Dq]PEPT[VE|PM|NN|Di]EEN[YN]HPSDE[SK]
もしこれが未知の3つの修飾を持つ本物のペプチドだったとしたら、曖昧な解にたどり着くのは至難の業でしょう。せっかくの大会なのだから、何かヒントがあるといいかもしれませんね。この3つの解答をしばらく凝視してください。すると、2つ目の解答の真ん中に「PEPTIDE」という文字があることに気づくかもしれません。これは偶然とは思えない。解答の右にある英単語がわかりにくいのですが、左側はすべてPHqDS[PH][Dq]で始まります。qDまたはDqをOの字で代用すると、PHOSPHOPEPTIDEとなります。
最初の説明文には、シーケンスには標準以外の文字が含まれていると記載されていたので、その点から色々と推理をしました。qDの質量は243.08です。これと各アミノ酸残基の質量の質量差を見てみると、チロシンでは80という値になります。そこで、修飾 された残基の一つがpYであり、これは配列中でOと表すと良いかもしれない、という仮説を考えました。
この仮説を元に話を進めてみます。Distillerのpreferences→”Sequence Tag/Denovo”タブにて、Phospho (Y)をvariable modificationに追加します。再びde novo計算をすると、最もスコアが高い答えが
PHYSPH[WG|SR|NE|Dq]PEPTPMEENRi[GC]HFENT
となります。PHOSPHOの2番目のOはなぜかアルゴリズムがYではなくDqを選択したままで、PEPTIDEもちょっと変わってしまいましたが、右辺がどうやらENRICHMENTのようである事がわかりました。そしてさらに今回の結果から修飾に関するさらなる推測も成り立ちます。修飾はおそらく最も一般的に遭遇する2つの修飾でしょう:GCはカルバミドメチル化されたCysと質量が同じで、Fは酸化Metと似ている事がよく知られています。そこで、fixed modificationとしてCarbamidomethyl(C)を、variable modificationとしてOxidation (M)を加えて再検索をすると、これまでで最高得点の解答になります。
PHqDSPHYPEPT[VE|PM|NN|Di]EENRiCHFENT
適切な条件で正解を導くのは難しいようで、最後は無理やり絞り込み条件を与えてみます。toleranceの値を狭め、Phospho(Y)をfixed modificationとしたところ、正解のペプチド配列らしきものとなりました。
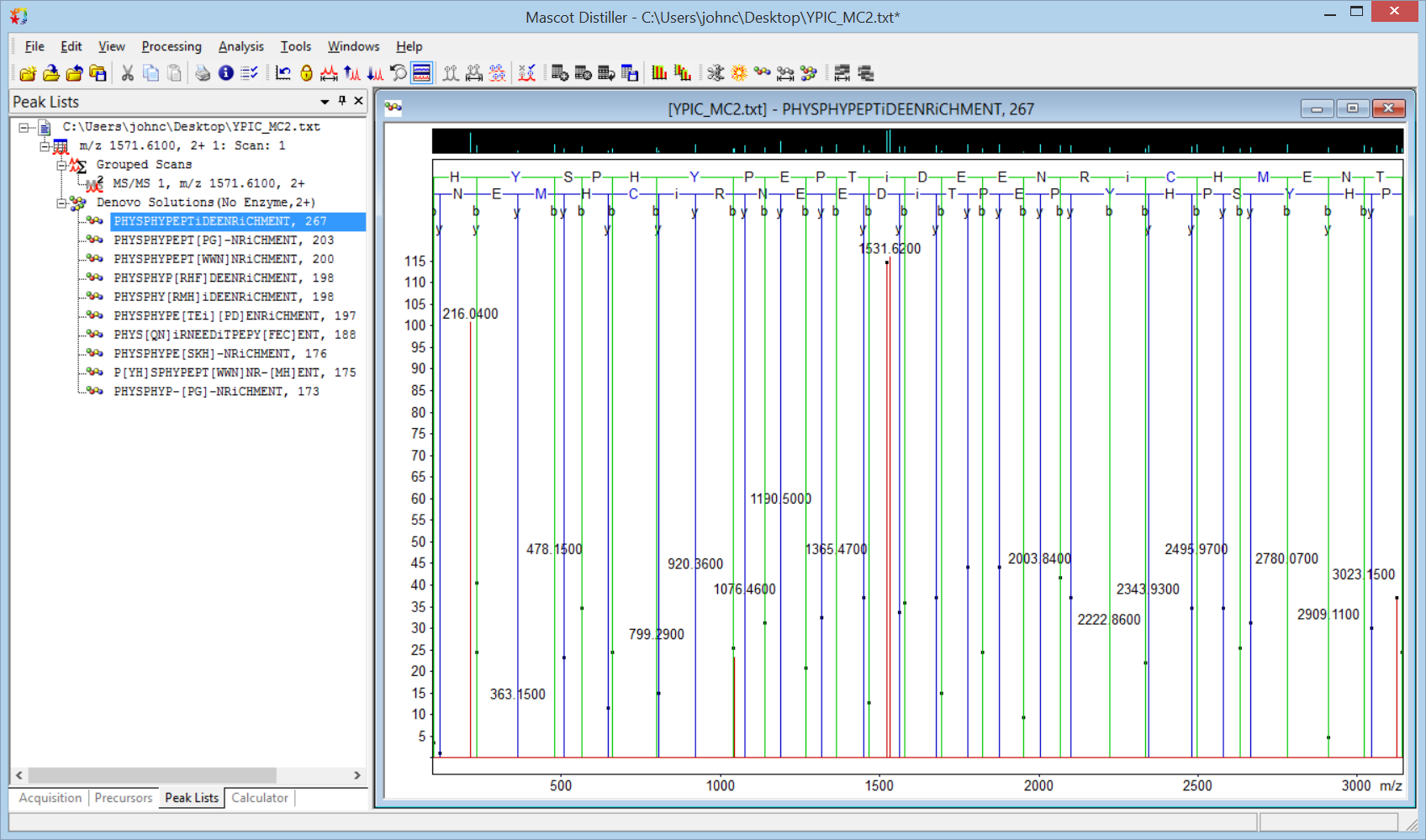
訳者 補足
筆者が楽しく推測している様子が伝わってくる記事でした。同時にde novo sequencingの難しさも実感しました。
Keywords: de novo, interpretation, Mascot Distiller, tutorial