シングルセルのペプチドフラグメンテーションスペクトル
装置の高感度化やサンプル前処理の最適化に伴い、シングルセルプロテオミクス研究は年々増加しています。最近、Journal of Proteome Researchに掲載された論文、「Features of Peptide Fragmentation Spectra in Single-Cell Proteomics(シングルセルプロテオミクスにおけるペプチドフラグメンテーションスペクトルの特徴)」では、データベース検索によるタンパク質やペプチドの同定にまつわる潜在的な課題を紹介しています。
Boekwegらは、バルクサンプル、2ngおよび0.2ng容量に分けられた少量サンプル、または単一のHeLa細胞からのペプチドのフラグメンテーションスペクトルについて、計算科学的なアプローチで調べました。シングルセルはnanoPOTS法で調製しました。この論文のキーポイントについて以下に紹介します。
ピーク強度の圧縮:著者は論文の中で、 「シングルセルのデータのスペクトルの大部分はピーク強度の”圧縮”が顕著で、シグナルとノイズの境界が曖昧になっている」と指摘しています。 ピーク強度の圧縮は2つの指標で測定され、このうちの1つである「T/M」というのはS/N比に関連しています。Tはアノテーションされた上位3つのフラグメントピークの平均値、Mは、アノテーションされていないピークの中央値です。論文中の図3Bは、バルクデータのスペクトルのT/M を、シングルセルのスペクトルのT/Mで割った値です。 大半のシングルセルスペクトルのT/Mは低く、バルクデータよりもはるかに低いことが多いようです。
このピーク強度の圧縮はMASCOTへも影響を与えるでしょうか。MASCOTではまず入力データであるスペクトルデータを100Da単位に分割し、各分割単位の中で最も強いピークを選択します。そのピークがフラグメント質量の理論値にマッチングするかを見ますが、この時質量の誤差だけを見て行い、フラグメントの強度については一切予測しません。続いて、各分割単位の中で先ほど選んだものの次に強度の大きいピーク、その次、と選んでいき、分割単位につき上位10個のピークまで解析対象とします。分割単位で選別する入力データ数を増やしていきながら、スコアの向上が止まってしまうまで検索対象ピークの選別対象の拡張を繰り返していきます。 また、マッチしたピークの強度の合計と、選択されたピーク全体の強度の合計のバランスをとるペナルティ項を備えています。すべての分割単位におけるフラグメントピークに対してノイズピークが圧倒的に多くないか、その強度の和を比較して、ノイズの方が目立って多い状態であればペナルティを与える、即ちマッチングスコアを下げる仕組みです。
MS/MSピークの数:2つ目の重要な発見は、シングルセルのスペクトルがバルクのスペクトルに比べてY-イオンピークの数が少ないという事です。Boekwogらはこのことを定量化するため、検索エンジンから出力された各ペプチド配列のyイオン数を数えバルクとシングルセルで比較しました。論文のFigure 2はyイオンの増減を図示したものですが、少し不思議な点があります。バルクデータで最もアノテーションされたyピークを持つペプチドが、シングルセルデータでは減りも最も多いという事です。しかしこの原因が検索に使用したデータベース特有の問題であるかは不明です。異なる検索エンジンを使用した場合別の結果になるかもしれませんし、ピーク抽出の手法に依存した問題かもしれません。
また著者は「バルクデータの典型的なスペクトルでは平均ピーク数が682であったが、この数と比較すると、2ngでのスペクトル数104、0.2ngでのスペクトル数86、シングルセルスペクトル98と他のデータではピーク数が圧倒的に少なくなる」と述べています。 しかしこの比較は少し論点がずれています。長さNのペプチドは N個のbイオンとN個のyイオンがあるので、完全にフラグメント化すると合わせて2N個のフラグメントの質量パターンが生成されます。もちろんニュートラルロスや複数電荷の状態もあり得ますが、そういったピークはペプチドの同定には不要なノイズです。逆に言えばフラグメントのピーク自体がスペクトルになければ、これらのその他のピークがどれだけあっても意味がありません。(訳者注:すなわちピーク数が2Nを超えている状態であれば、スペクトルに含まれるピーク数の多少を議論する事にあまり意味はない、という意図だと思われます)
より適切な表現としては、「シングルセルのスペクトルは内部的に一貫しているがバルクのスペクトルとは明らかに異なる。」という事になるでしょうか。 これは、バルクデータのPSM(Peptide Spectrum Matches)とシングルセルデータのPSMのコサイン類似度を比較することで確認きます。この事が示すのは、シングルセルデータの解析にはシングルセルデータに特化したスペクトルライブラリを構築すべきだということです。特に、フラグメンテーションパターンを予測するPrositのような機械学習ツールのトレーニングにスペクトルが使用される場合、注意しなければなりません。
同定基準値:S/Nが悪くなり検出されるフラグメントピーク数が少なくなれば、マッチングスコアは低くなります。この点はBoekwogらも指摘しています。Mascotには、各queryに固有の2つの同定基準値があります:identity thresholdとhomology thresholdです。Identity thresholdは 単純に試行(候補ペプチド)の数から導かれ、スペクトルの質に影響されません。一方homology threasholdは、候補ペプチドのスコア分布に基づく統計的モデルから算出され、入力データが候補ペプチドとどのようにマッチするのかという事に依存しています。MASCOTでは通常、よりスコアが低いhomology threshold を同定基準として採用していますが、homology threshold を計算した結果identity threshold より高くなる場合はidentity threshold が同定基準値となります。今回のようにMASCOTの同定結果をそのまま適用してもうまくいかない場合の1つの解決策として、Percolatorのような機械学習ツールを使用して感度の向上(訳者注:この場合は同定ペプチド数の増加)を図る事が挙げられます。
MASCOTの検索例:この件についてより詳し確認するため、MassIVE,MSV000087524のシングルセルのrawデータの1つをダウンロードしMascot Distillerでピーク抽出をし検索をしました。 rawデータはThermo Orbitrapで測定されたもので、MS/MSスキャンはセントロイドデータです。ピーク抽出の条件に関する設定はデフォルトでも問題ありませんが、先月のブログ記事「Defaultとprof_prof どちらが良いか?」でも述べたように、prof_prof設定を使用することで結果が少し改善されます。
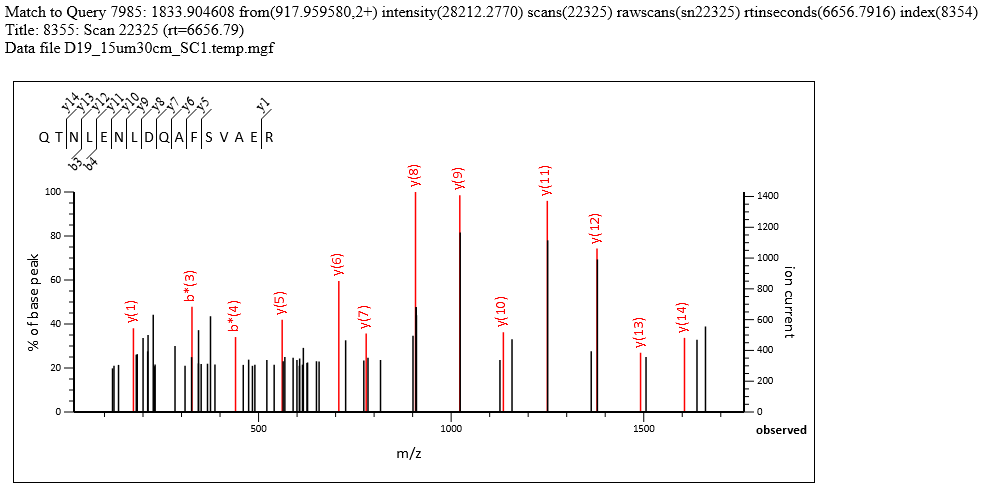
上記データを検索したこちらの結果では 9,902件のqueryのうち、1% FDRで同定基準を超えるPSMが2007、配列は1726でした。検索パラメーターはHeLaサンプル検索では典型的なセットを利用していて、プリカーサーとフラグメントの誤差範囲はともに20ppm以内です。高いノイズピークの存在にもかかわらず、多くの高スコアのマッチが得られました。 例えばquery7985のQTNLENLDQAFSVAERなどです(下図)。T/M指標は3.0前後で、上位3つのピーク強度の平均は1000~1100程度、一方ノイズのメジアンは300~350程度です。 可能性のあるすべてのマッチをラベル付けすると、強度30%以上のピークのほとんどがアノテーションされます。
ピーク強度が圧縮されているもう一つの例は、query 6066 のAGPGTLSVTIEGPSKへのマッチングです。このケースでは、MASCOTが行っている100Da分割単位でピックアップするピーク数を増やしていく操作を進め、よりフラグメントのピークを見つけていく必要があります。 しかしこの例では明確なシーケンスラダーを見つけながら、しかもプリカーサーの誤差許容範囲内には他の候補ペプチドが現れません。ほとんどのyイオンには+1Daのピークが隣接しています。 これは同位体のピークですが、Distillerのprof_profのピークピッキングオプションを使うと除去することができます。
Percolator を有効にして、使用するパラメータとしてはMascot Server 2.8で導入されたデフォルトのfeature(訳者注:考慮するパラメーター)セットに加えRTも使用するように設定して検索をしてみます。すると1% FDR では 2303の有意な PSM と 1991の有意な配列が得られます。そしてこれらペプチドから571個のタンパク質がヒットしました(protein FDRは3.2%)。このように、query数が少数の割にはかなり良い結果が得られました。
実験の目的が細胞の不均一性を調べる事である場合、technical replicatesという概念がそもそも存在しないという事になります。シングルセルの測定の場合、各細胞は厳密に一度だけ測定されるからです。 感度を上げ同定されるタンパク質の数を増やすための最もインパクトのある方法は、おそらくtimsTOFやFAIMSのようなイオンモビリティーによる分離を適用する事です。イオンモビリティーの適用は、ペプチドデータの混合を防ぎスペクトルの質が向上します。 Boekwogらも同じ問題に着目しています。
この研究に使用されたシングルセルのrawファイルに含まれるquery数は20,000以下ですが、現在弊社公開サイトでは一度の検索にかける事ができるquery数の上限を上げており、20,000 queryでも検索が可能です。興味がある方は是非実際に検索していろいろと試してください。 またMascot Distillerのトライアルライセンスをご希望の方は弊社までご連絡ください。
Keywords: fragmentation, scoring, single-cell proteomics