チュートリアル:最適なMS2PIPモデルの選択
Mascot Server 3.0は、フラグメント強度の予測値を使用してデータベース検索結果をより良いものにすることができます。予測値はMS2PIPによって計算されます。Mascotには一般的な装置タイプに適したMS2PIPのモデルが予め準備されています。このチュートリアルでは、装置と実験に最適なモデルの選択方法について説明します。
MS2PIPとは?
MS2PIPは、ペプチド配列、電荷状態、アミノ酸への修飾内容からMS/MSフラグメンテーションスペクトルを予測するツールです。最も一般的なフラグメントイオンの強度(b、y、b++、y++)について予測します。
予測は、Mascotのような従来の検索エンジンが提供する大量のペプチドスペクトルマッチングの結果からモデルをトレーニングすることで行われます。MS2PIPはオリジナルの論文で説明されているように、ランダムフォレスト回帰の手法を使用しています。そしてその後の論文でも発表されているように、さらに改良が加えられています。
Mascotにおいてピーク強度予測を行う目的
Mascotの確率に基づくスコアリングでは、ピーク強度を使用してシグナルとノイズを区別します。スペクトルは100Daのウィンドウに分割され、各ウィンドウでは最大10個の高強度ピークがマッチング・スコアリングのために選択されます。Mascotは、イオンシリーズの異なる組み合わせを使用して、観測された強度の最大量を説明することを目的としています。ただし、ペプチドのマッチングスコアはあくまでもフラグメントの質量一致具合に基づいています。ピーク強度は間接的にスコアに影響しますが、Mascotのイオンスコアに対して直施設的に影響を与えません。
新しい仕組みで検索結果を再スコアリングする時、MASCOTは予測保持時間情報と共にフラグメントの予測強度を使用します。特に、予測スペクトルと観測スペクトルの相関に着目します。適切なMS2PIPモデルを選択した上で利用する事で、誤ったマッチングのスペクトル相関は分布が広がり相関係数が0に近くなる一方、正しいマッチングの相関係数は1.0に近くなります。このようなクラスタリングは、Percolatorの半教師付き学習に最適です。
MS2PIP の使用方法
Mascot Server 3.0 は MS2Rescore と統合されており、MS2PIP はそのコンポーネントの1つです。本来MS2PIP は使用前にトレーニングする必要がありますが、Mascot Server 3.0 には13の事前トレーニング済みモデルが付属しています。データベース検索が終了後、結果レポートのフォーマットコントロールからモデルを選択し結果の最適化を実行してください。

適したモデルを選択する方法
サンプルの測定に関する情報が必要で、以下に具体例を挙げます。使用した装置はどのメーカーのどのモデルか。アイソバリック定量(TMT/iTRAQ)、代謝定量(SILAC)のサンプルなのかどうか。ペプチドはトリプシン消化またはセミトリプシン消化、または内因性なのか。サンプルはリン酸化などの特定の修飾について濃縮されているかどうか、など。
続いて、MS2PIPのヘルプページをご覧いただき、ご使用の装置と切断処理(消化酵素設定)に最も適したモデルを選択してください。
概ねのケースにおいて、CIDモデルから検討を始めるのが良いでしょう。このモデルは、多数のCID装置から得られた共通のスペクトルを厳選した、高品質なNIST CID Humanスペクトルライブラリをトレーニングデータとして利用しています。従ってMS2PIPモデルは、あらゆる種類の装置におけるCIDフラグメンテーションの一般的なルールを捉えるようにトレーニングされていると言えます。
iTRAQ ラベルを使用した場合、iTRAQ モデルまたは iTRAQphospho のいずれかを選択してください。これらのモデルは Orbitrap スペクトルでトレーニングされていますが、ラベルは装置タイプよりもはるかにペプチドのフラグメンテーションを変化させるため、これらのモデルは Orbitrap 以外の装置にも適しています。
TMT ラベルを使用した場合は、TMT モデルまたは CID-TMT のいずれかを選択してください。TMT ラベルの有無は、正確な MS/MS 予測における重要な特性です。このモデルはOrbitrapデータでトレーニングされていますが、Orbitrap以外の装置にも適しています。CID-TMTモデルは、MS3を使用して取得したスペクトルの特殊なケースです。
ThermoのOrbitrapを使用している場合は、HCDモデルのいずれかを選択するのが最適です。HCD2019はトリプシン消化ペプチドのデフォルトとして適しており、Immuno-HCDまたはHCD2021はトリプシン消化ペプチド以外のペプチドに適しています。Sciex 装置を使用した場合、TTOF5600 は Sciex 5600 および 6600 装置の両方に対して良好な予測を提供します。Bruker timsTOF を使用した場合は、timsTOF2023 または timsTOF2024 のいずれかを選択してください。
モデルを選択した後、重要な指標は予測スペクトルと観測スペクトルの相関です。機械学習品質レポートに移動し、”Rescroing features”タブを開きます。「MS2PIP model performance」は予測スペクトルと観測スペクトルの相関におけるピアソン係数のヒストグラムです。featureの「spec_pearson_norm」に現れます。相関係数の中央値が0.85以上であれば良好で、ヒストグラムの大部分が0.8~1.0の範囲に集まっているのが理想的です。
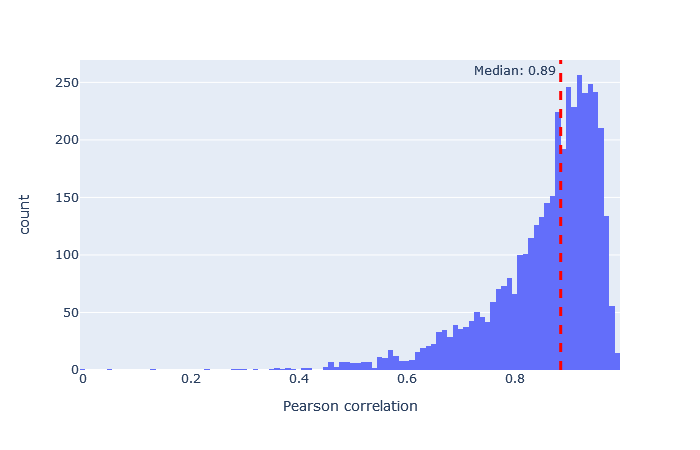
中央値の相関が低い場合、非常に広い範囲に広がっている場合、あるいは複数のピークがある場合は、別のモデルを試してください。 適合したモデルの例と適合しなかったモデルの例は、以下に表において示します。
実験条件に適したモデルが見つかれば、以降の解析でこの手順を繰り返す必要はありません。デフォルトとして、Daemonパラメータエディタにモデルを保存するか、検索フォームのデフォルトとしてクッキーに保存して次回以降も同じモデルを適用してください。
予測スペクトルはどのようなものですか
MS2PIPは、bイオンおよびyイオンに対するm/z値の短い配列と、各イオンに対する予測強度を返します。予測強度は、(0.0, 1.0)の範囲の値を取るよう、正規化されています。現在Mascotで予測スペクトルを表示する事はできませんが、公開されているMS2PIPのサイトを使用して、単一ペプチドに対する予測をグラフ表示することができます。
例えば、検索例の クエリ4220であるDAGTIAGLNVLR、2+の予測スペクトルを取得したいとします。オンラインMS2PIPサーバーにアクセスするには、https://iomics.ugent.be/ms2pip/#runを開きます。(Mascotではiomics.ugent.beでホストされているサーバーを使用しませんが、このサイトではMascotに同梱されているものと同じバージョンのモデルを実行しています。)該当するfixed/variable 修飾とモデルを選択します。今回の例ではCIDを選択します。続いて以下に記す行をmy_peptide.peprecとして保存します。これはペプチドの特性情報です。
spec_id modifications peptide charge DAGTIAGLNVLR_2 - DAGTIAGLNVLR 2
それをPEPRECファイルとして選択し、出力フォーマットをCSVに設定してから”start”をクリックします。しばらくすると結果ページにリダイレクトされ、予測スペクトルのプロットとCSV形式での予測が表示されます。
Mascotで観測されたスペクトルと、以下に示す予測スペクトルを比較します。

ペプチド配列 DAGTIAGLNVLR, 2+の観測スペクトル
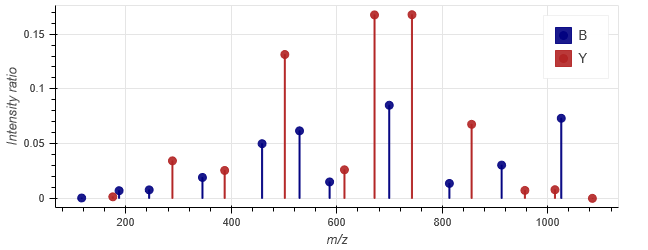
CIDモデルを使用した、ペプチド配列DAGTIAGLNVLR, 2+の予測スペクトル
悪くない結果ですね!存在するbイオンは、相対強度が観測値と非常に近いです。yイオンでは、Mascotによって注釈付けされたピークは、bとyに加えてaイオンとして示されている事があります。a6/y4やa8/y6などのいくつかのピークは、フラグメント許容範囲内(この場合は0.6Da)で同じ測定データのピークと一致しており、Mascotはそれらをaイオンとして注釈付けしています。しかし、501.3144 m/z における a6/y4 の相対強度は予測値とほぼ一致しており、a8/y6 の相対強度も同様です。
少数のスペクトルについて目視確認を行うのは明らかに非現実的と言えます。モデル選択を評価するには他の方法が必要です。
誤ったモデルを選択した場合、どのようなことが起こるか
適合性の悪いモデルを選択した場合どうなるでしょうか?幸いなことに、結果が適用前より悪くなることはほとんどありません。例えば、モデルがすべてのペプチドに対して同じフラグメンテーションパターンを予測する場合、ターゲットとデコイのスペクトル相関は極端にばらつきます。結果的にPercolatorは、正しいマッチングと誤ったマッチングを区別するために、この要素を使用しない事になります(正解と不正解を分離する情報を提供していないとみなされるため)。
トリプシンモデルを使用して、トリプシン消化以外のペプチドのフラグメンテーションスペクトルを予測することは可能ですが、正確ではない場合があります。一方、セミトリプシンまたは内因性ペプチドでトレーニングされたモデルは、トリプシンペプチドのスペクトル予測では通常、良好なパフォーマンスを示します。
以下の表は、Mascotにセットされている各モデルを適用した場合のピアソン相関係数をまとめたものです。データセットは、8月のブログ記事で使用したCPTAC酵母サンプルの検索結果で、Thermo LTQ-XL-Orbitrapで取得したものです。
ペプチドFDRが1%の時の同定ペプチド数も示しています。同定ペプチド数については、Percolatorを使用するものの、DeepLCもMS2PIPモデルも選択しない場合となります。2740の同定ペプチドが得られます。表中の同定ペプチド数は、純粋にMS2PIPにより増加がもたらされているケースです。
| Model | Sig. unique sequences | Median correlation | Histogram | |
|---|---|---|---|---|
| CID | 3514 (+28%) | 0.89 |
|
|
| CIDch2 | 3514 (+28%) | 0.89 |
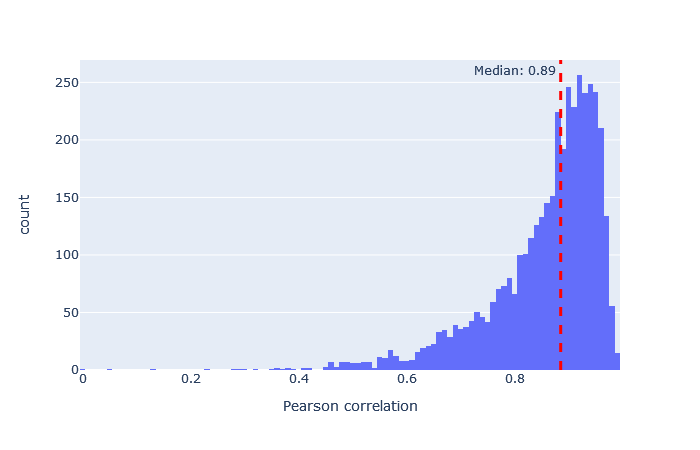
|
|
| TTOF5600 | 3311 (+21%) | 0.65 |
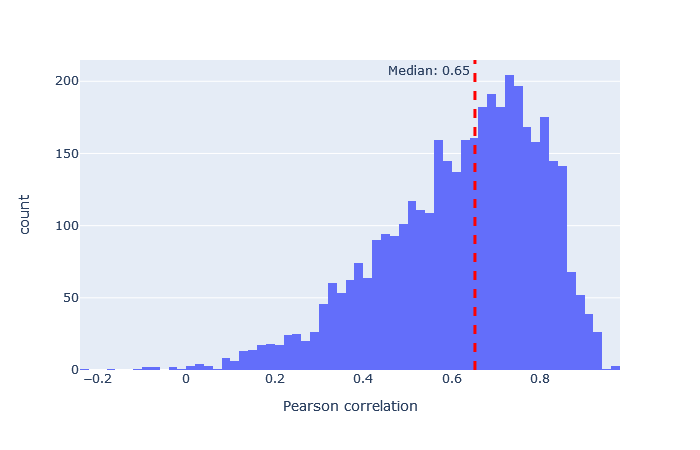
|
|
| HCD2019 | 3257 (+19%) | 0.56 |
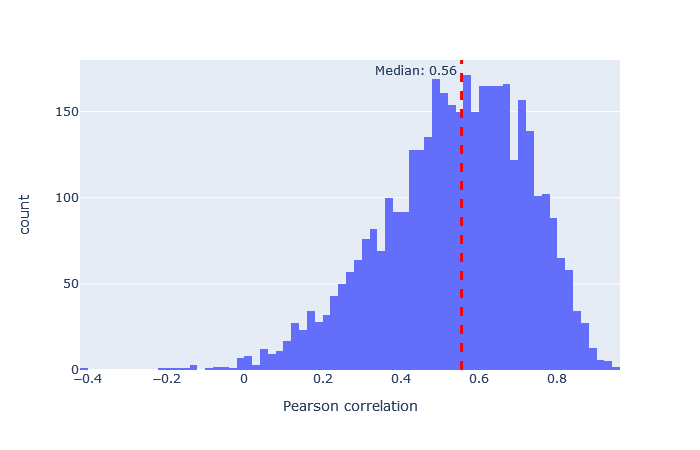
|
|
| HCDch2 | 3257 (+19%) | 0.56 |
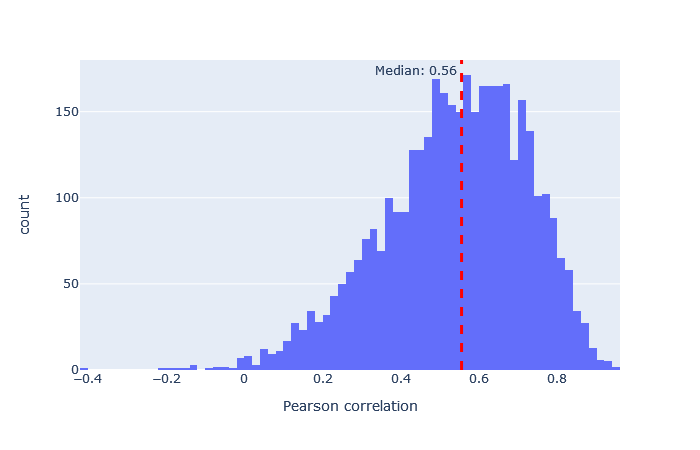
|
|
| HCD2021 | 3157 (+15%) | 0.54 |
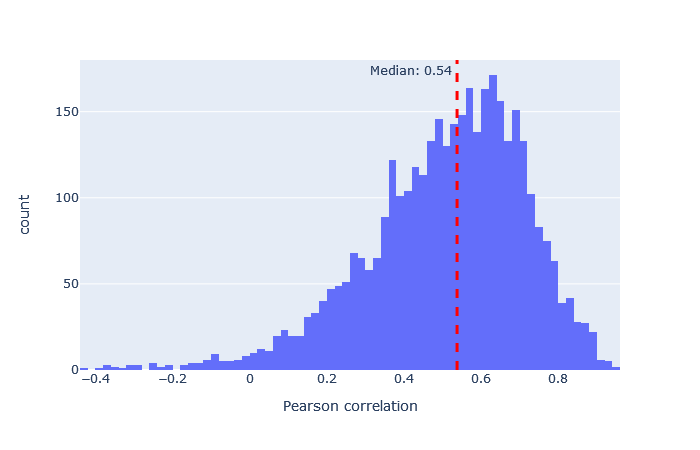
|
|
| timsTOF2024 | 3149 (+15%) | 0.69 |
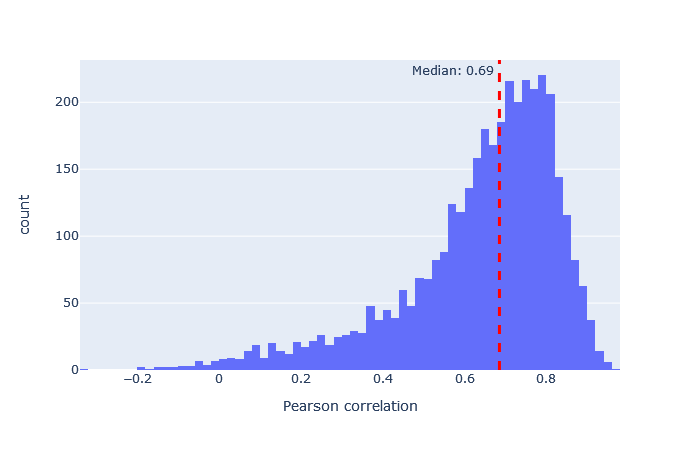
|
|
| TMT | 3130 (+14%) | 0.13 |
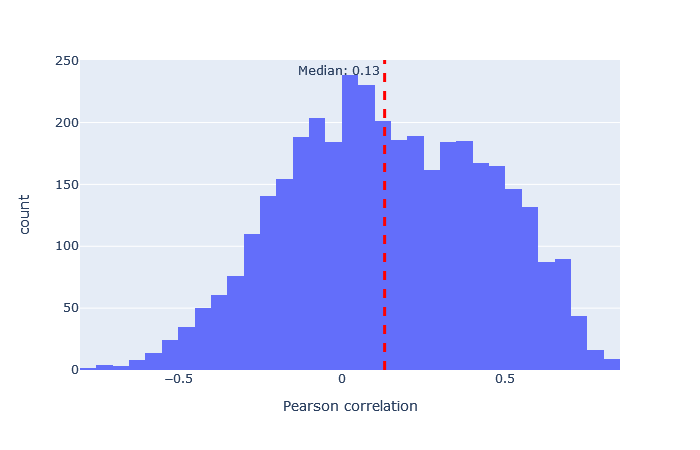
|
|
| timsTOF2023 | 3121 (+14%) | 0.69 |
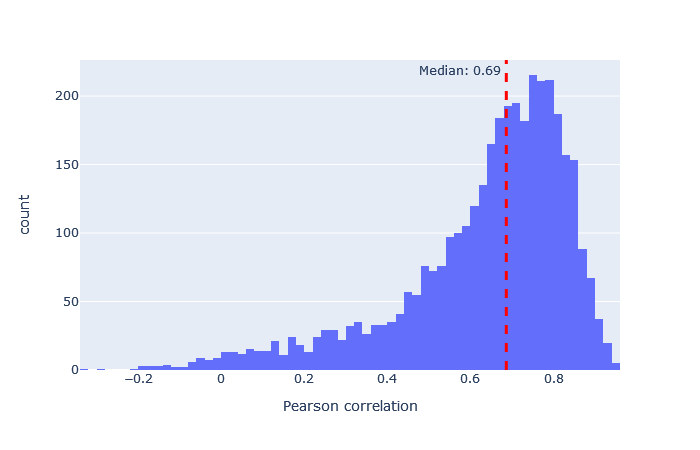
|
|
| CID-TMT | 3119 (+14%) | 0.46 |
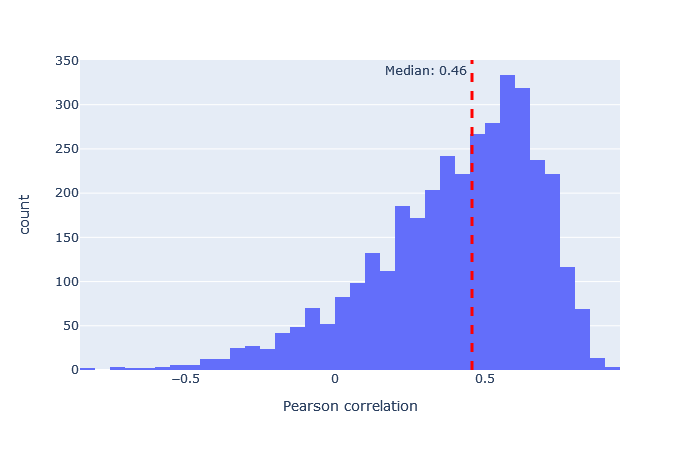
|
|
| Immuno-HCD | 3053 (+11%) | 0.53 |
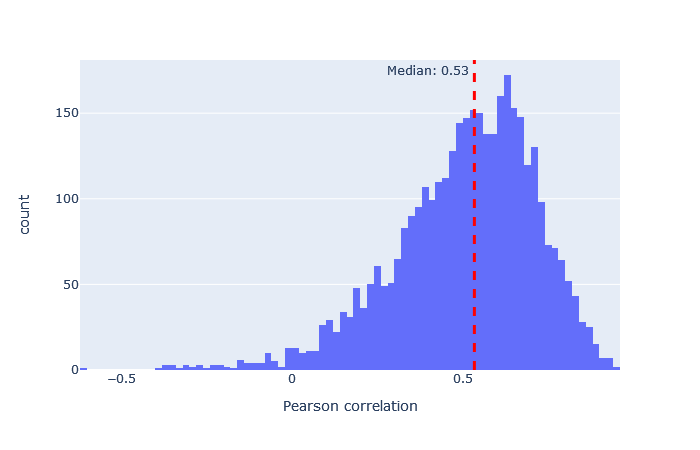
|
|
| iTRAQ | 3015 (+10%) | 0.20 |
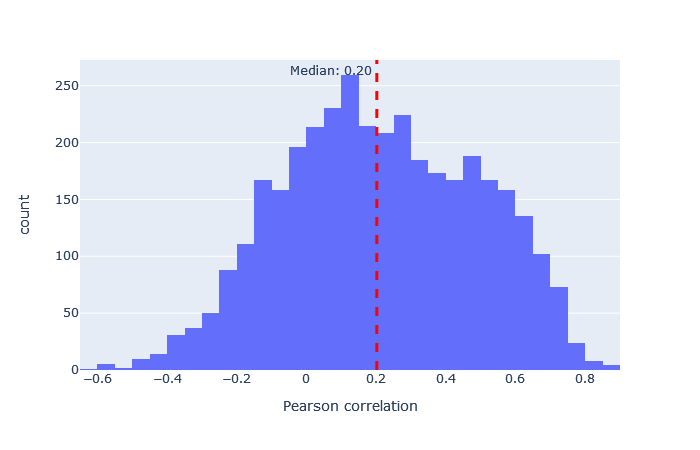
|
|
| iTRAQphospho | 2939 (+7%) | 0.12 |
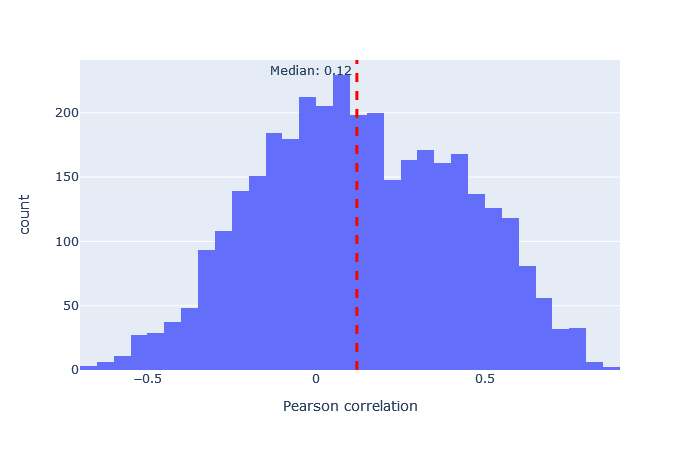
|
非常に適合度の悪いモデルでも、配列FDRが1%の場合にはユニークな配列の数をわずかに増加させています。例えば、iTRAQphosphoの相関係数の分布はほぼ0を中心に左右対称に分布しており、予測は主にランダムであることを示していますが、予測が十分に正確なため、分布がわずかに右に偏っているペプチドが少数存在します。この違いは、Mascotで計算されたコア機能と併用した場合、Percolatorが正しい一致と誤った一致を区別する要素として使用するには十分です。 この表からも、最悪のモデルと最良のモデルの適合度の差は非常に大きいため、常に最良の適合モデルを選択すべきであることは明らかです。
Keywords: machine learning, MS2PIP, Percolator