複数の前駆体ペプチドを選別する : Mascot DIAにおける前駆体ペプチド検出
私たちは、DIAデータの分析のための汎用的なスペクトル中心方式のソリューション「Mascot DIA」の開発を進めています。このソリューションのプレビューは、当社の ASMS 2025 ブレックファストミーティングで発表されました。その時のスライドは無料でダウンロード可能です。 先月のブログ記事(英語、日本語)では、DIAデータに対するスペクトル中心方式の検索アプローチの概要を説明し、より一般的なペプチド中心型アプローチとの比較を行いました。スペクトル中心方式のアプローチにおいて不可欠な要素の一つが正確な前駆体ペプチドの検出です。Mascot DIAではこれをどのように実現しているか、以下に説明いたします。
Mascot DIAにおける前駆体ペプチド検出は、以下の図に示すようにMascot Distillerで行われます。
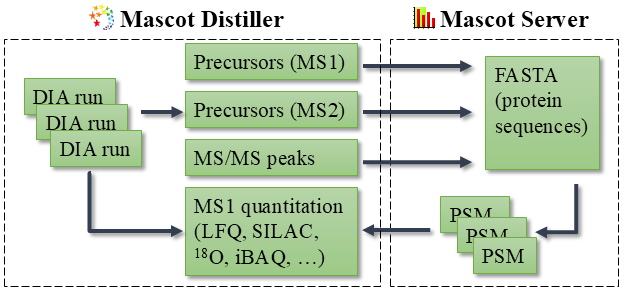
図1:Mascot DIAのワークフロー。前駆体ペプチド検出とピーク抽出はMascot Distillerで行われ、ピークリストはMascot Serverデータベース検索で照合されます。検索結果はMS1をもとにしたの定量計算のため、Mascot Distillerにインポートされます。
Mascot Distiller 3におけるDIAデータの前駆体ペプチド検出
Mascot DistillerにおけるDIAデータの前駆体ペプチド検出は2段階のプロセスです。まず最初にソフトウェアはサーベイスキャンを検索し、サーベイスキャンで十分な前駆体ペプチドを検出できない場合、MS/MSスペクトル内から前駆体ペプチドを検索する新しいアルゴリズムを使用します。
このアプローチの主な利点は、前駆体ペプチド検出(そしてペプチド同定)が、実験的あるいは計算で導かれた前駆体ペプチド質量と保持時間のライブラリに依存しない点です。これにより、Mascot Serverでサポートされる任意の翻訳後修飾を有するペプチドや任意の酵素消化によって得られたペプチドを検出できる可能性があります。
ステージ1:サーベイスキャンを検索
標準的なDDA実行と同様、Mascot DistillerでのDIA実行における前駆体ペプチド検出はMS1サーベイスキャンから開始されます。サーベイスキャンは、DIA測定で使用された分離ウィンドウの幅で分割され、それぞれのウィンドウ内で選択された前駆体ペプチドの質量が対応するMS/MSスキャンに割り当てられます。このプロセスでは、DDAデータの前駆体ペプチド検出に使用されるのと同じアルゴリズムを使用します。
DIAデータに関する解析では、すべてのスキャンがキメラ(複数の前駆体ペプチドが混合している)状態であることを前提とします。したがって、Mascot Distiller が特定の分離ウィンドウに対してサーベイスキャン内に複数の前駆体ペプチドを見つけられなかった場合、さらにMS/MSスキャン内で追加の前駆体ペプチドのピークを探索するように切り替えます。
この機能は、DIA解析をサポートするために Mascot Distiller 3 に新たに追加されたものです。
ステージ2:MS/MSスキャンを検索
DIAデータセットのMS/MSスキャンには、フラグメンテーションを起こしていない前駆体ペプチド由来のピークがしばしば見られます。そのため対処法の一つとして、MS/MSスキャンの分離ウィンドウ領域を調べ、そこから未分解の前駆体ペプチドの候補を抽出する方法が考えられます。しかし検証の結果、この方法ではさまざまな理由からあまり良い結果を生み出さない事が分かりました。例えば、前駆体ペプチドがサーベイスキャンに存在しないケースというのは本質的に低強度の前駆体ペプチドであることが多く、MS/MSスキャンでもやはり未分解の前駆体シグナルが検出されにくく見落とされたり、他のノイズに埋もれてしまう可能性が高いというケースがありました。
もうひとつの問題は、rawデータがセントロイド形式で保存されている場合です。これは、Thermo製のOrbitrapのような装置からのデータにしばしば見られます。このような場合、Mascot Distiller はフラグメントイオンの電荷状態を検出できません。検出するにはスペクトルをプロファイルデータに戻す「アンセントロイド処理」が必要となり、処理時間が大幅に延びてしまいます。
私たちは別の戦略をとることにしました。Mascot Distiller では新しいアルゴリズムを用いて、補完的なイオンのペア(たとえば bイオンとyイオン)の情報を使って、分離ウィンドウ内の前駆体ペプチドの質量を推定する方法を採用しました。以下のような手順で解析を行います。
- MS/MSスペクトルからフラグメントイオンのペアの質量を合計します。これにより、潜在的な前駆体ペプチドにプロトンが追加された値(MH+)がわかります。追加のプロトンの質量を差し引いた後、前駆体ペプチド質量が分離ウィンドウ内に収まるかどうかを確認します。問題なければ前駆体ペプチドの可能性があるとして、「ペプチド質量情報としての妥当性を検討するべきリスト」に加えます。
- 質量が前駆体ペプチドのウィンドウ幅を超えていた場合、処理設定で指定されたデフォルトの電荷状態の範囲内で(例:2+、3+など)変換してみます。適用の結果問題なければ、、「ペプチド質量情報としての妥当性を検討するべきリスト」に加えます。
- 手順1と2を、MS/MSスペクトル全域で繰り返します。
- 「ペプチド質量情報としての妥当性を検討するべきリスト」の候補ペプチドについて、異なるフラグメントイオン対によって観測された回数とスペクトルカバー率でランク付けし、上位N個の前駆体ペプチドを選択してスペクトルに割り当てます(Nはデフォルトで10です)。
このルールは、Dancikら[1]が提案したde novo sequencingアルゴリズムに類似しており、以下の利点があります:
- MS/MSスペクトルデータ駆動型であるため、前駆体ペプチド質量と関連する保持時間のライブラリを必要としません。
- ペプチドに存在する可変修飾や、消化酵素の種類に影響を受けません。
- セントロイド化されたデータからでも前駆体ペプチド質量と電荷を決定できます。
- フラグメント効率にほぼ依存しません – ペプチドが十分に断片化されて補完的なイオンをいくつか生成できれば、前駆体ペプチド質量を決定できます。
- b-とy-、c-とz-など、任意の補完的なイオンシリーズに対応します。
MS/MSに基づく前駆体ペプチド検出の例
DIAデータセットにおけるマッチングの例です。MS/MSスキャンから前駆体ペプチドm/zと電荷状態が次のように決定されています。マッチング結果は図2に示されています:
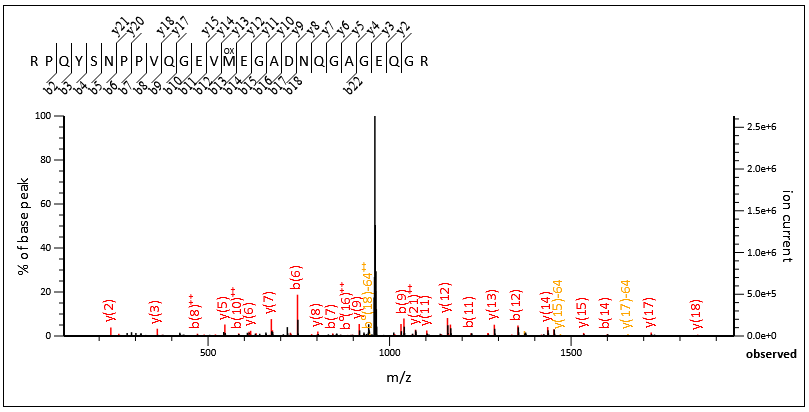
図2: RPQYSNPPVQGEVMEGADNQGAGEQGRのMS/MSフラグメンテーション
ご覧の通り、ペプチド全体に良好なカバレッジが得られています。1150-1750の領域を拡大すると、複数の補完的なイオン対が存在します:b12+y15、b13+y14、b14+y13:
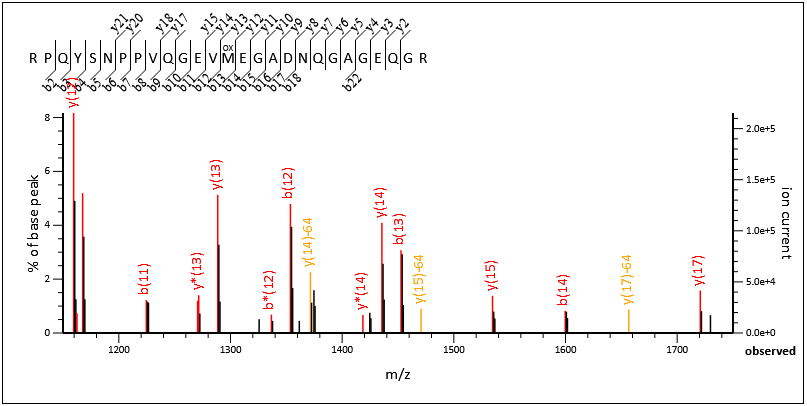
図3: 1150-1750の拡大領域に存在する複数の補完的なイオン対
これらの対の計算された質量と電荷は、以下の表1に示されています:
| Pair | m/z b- ion | m/z y- ion | MH+ plus proton | 1+ | 2+ | 3+ |
|---|---|---|---|---|---|---|
| b12+y15 | 1353.652 | 1534.6479 | 2888.3 | 2887.293 | 1444.15 | 963.1024 |
| b13+y14 | 1452.7212 | 1435.5948 | 2888.316 | 2887.309 | 1444.158 | 963.1078 |
| b14+y13 | 1599.7555 | 1288.5535 | 2888.309 | 2887.302 | 1444.155 | 963.1054 |
表1: 3組の補完的イオンからの前駆体ペプチド計算例
このMS/MSスキャンにおける分離ウィンドウは956.687-964.687であり、前駆体ペプチドはm/z 963.1で電荷状態3+であれば条件を満たします。実際に、Distillerで計算された前駆体ペプチドはm/z 963.10696,電荷が3+であり、前駆体ペプチド誤差は1.37ppmです。MS/MSスキャンにおける分離ウィンドウ領域に、この前駆体ペプチドと一致しない強い未分解前駆体ペプチドピークが存在していますのでその点にご注意ください(図2参照)。図4は、MS/MSスキャンから同定された分離ウィンドウ内の異なる前駆体ペプチド質量に対するb- y-補完ペアの数を示しています。このケースでは、プログラムにて計算された前駆体ペプチドm/z 963.10696 3+は明確な外れ値です:
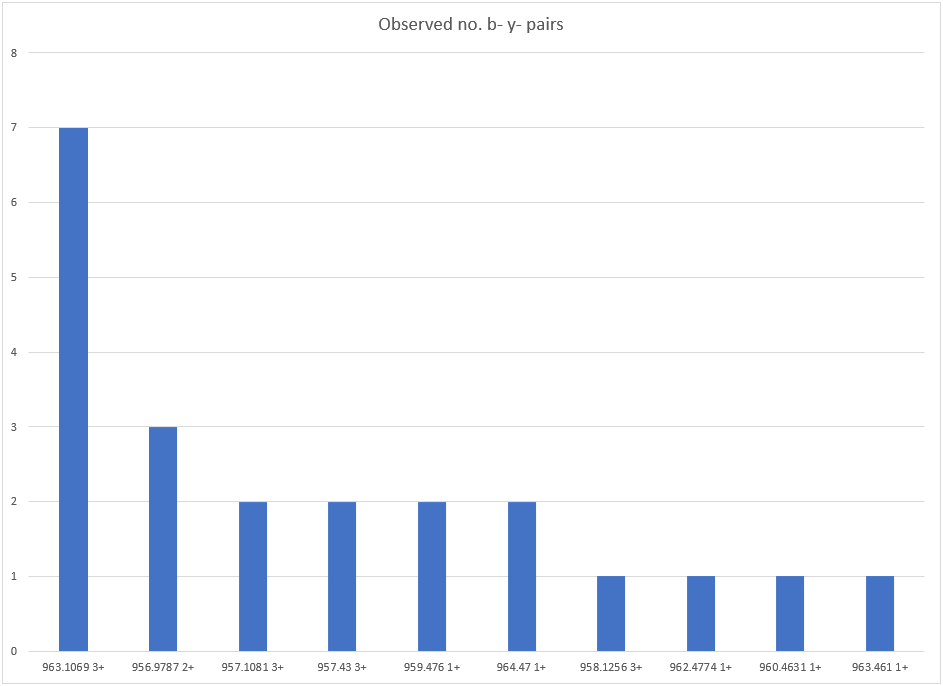
図4:MS/MSピークリストから同定された様々な前駆体ペプチドに対する補完b- y-イオンペアの数を示す棒グラフ。
Reference
- Dancik V, Addona TA, Clauser KR, Vath JE, Pevzner PA. De Novo Peptide Sequencing via Tandem Mass Spectrometry. Journal of Computational Biology 6(3/4):327-342. DOI: 10.1089/106652799318300
Keywords: chimeric spectra, DIA, Mascot Distiller, peak picking