Mascot Distiller で Thunder-DDA-PASEF データを処理する
ヒト白血球抗原(HLA)クラスIペプチドリガンド(HLAIps)は、ワクチンおよび免疫療法の開発における重要なターゲットとなります。HLAIpsは通常の質量分析法に基づくプロテオミクスでは同定が困難です。というのも、HLAIpsは短く、存在量が少なく、特定のプロテアーゼによって生成されるわけではないため、酵素特異性を考慮しない検索を行う必要があり、標準的なトリプシン消化酵素処理されたペプチドと比較して検索範囲が劇的に拡大してしまうためです。
こういった問題に対処するべく、timsTOF + LC-IMS-MSで最適化された測定方法として、Thunder-DDA-PASEF法 [1]が最近開発されました。1価のプリカーサーが頻繁に存在しペプチド長の分布幅が狭いHLA-Ipsの特性に合わせて、標準的なDDA-PASEFワークフローを改良したものです。この手法の名前は、1/K0イオンモビリティーおよびm/zチャネルに適用されるフラグメント分離フィルターの稲妻のような形状に由来しています。
DDA-PASEFファイルはMascot Distillerで処理可能であり、Thunder-DDA-PASEFデータも例外ではありません。検索空間が広すぎて本来は有意なマッチングが得られないHLA+非特異的な切断データの解析でも、MS2PIPおよびPercolatorを用いた機械学習による再スコアリング法を適用することでその一部を同定する事が可能となります。
解析例
この手法について検証するために、論文(PXD040385)に関連する JPOST リポジトリからいくつかのデータファイルを取得しました。半分は標準的な DDA-PASEF 法、残りの半分が Thunder-DDA-PASEF 法で測定されたデータです。残念ながらこのアーカイブには論文の図2を作に使用されたrawデータファイルが含まれていません。しかし、論文や補足データには記載されていない別の実験において、標準DDA-PASEF法とThunder-DDA-PASEF法で同一の血漿サンプルを比較した測定データのrawファイルがそれぞれ3つずつ含まれていました。私たちは、Mascot DistillerおよびMascot Server 3.1を使用して、これらのファイルを再処理し検索を行いました。
Mascot Distillerでの処理はtimsTOFファイル用に提供されているデフォルトのパラメーターをベースにしましたが1点変更を加えています。変更点は、処理オプションファイル内の
<precursorIonMobilityGroupTol>
という、プリカーサーのグループ化に対する1/K0許容誤差に関する値です。デフォルトでは0.026に設定されていますが、より厳密な許容誤差である0.01に変更しました。現在Distiller上から設定変更できませんが、設定ファイルをテキストエディタで開くことで編集可能です。Distillerの次期リリースではGUIから編集できるようになる予定です。
検索設定は論文の内容に基づいており、検索に使用したデータベースは論文で使用された生物種混合データベースと同じものを利用しています。酵素切断設定は非特異的な"None"を適用しました。結果はPercolatorのfeature標準設定、およびMascot 3.1に同梱されているMS2Rescoreパッケージの一部として提供されるMS2PIP(timsTOF2024モデル)を使用して再スコアリングされリファインメント(精緻化)しました。有意性の閾値は、1%のFDR(配列ベース)を適用しています。結果は以下表1の通りです。
| Significant sequences (1% FDR) | ||
|---|---|---|
| Isolation filter | Percolator | Percolator+timsTOF2024 |
| Standard | 3337 | 4937 |
| Thunder | 6378 | 8814 |
Table 1:標準およびThunderの分離フィルタ、timsTOF2024モデル 有り/無し で再処理を行った検索結果、同定ペプチド数(1% FDR、配列ベース)
この結果から、Thunder-DDA-PASEFファイルを使用すると、標準的な手法と比較して1% FDRにおける有意な配列数が大幅に増加することがわかります。またいずれの場合もtimsTOF2024モデルの予測を含めることで、有意な配列数が40%以上増加します。以降のすべての比較は、Percolator + timsTOF2024 の設定を使用して実施しました。
Thunder DDA-PASEF分離スキームは、HLAIpsの予想プロファイルに一致するペプチドイオンの捕捉を改善するための設計がされています。例えば、一価のペプチドをより多く含める事ができるようになっています。以下の図1では、標準およびThunder分離スキームにおいて有意なPSMの電荷状態分布の比較をしています:
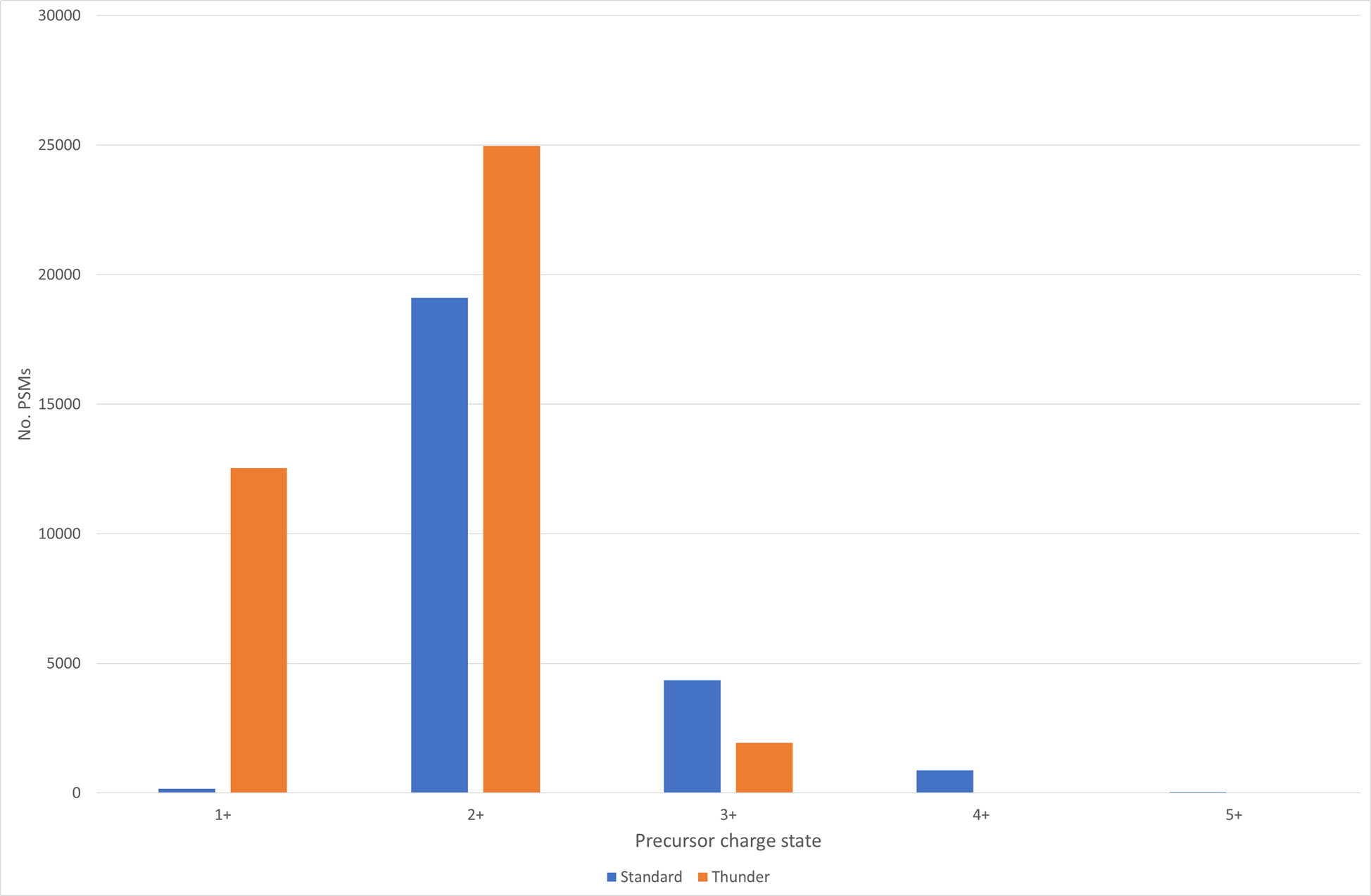
Figure 1: 標準的な方法 vs Thunder DDA-PASEF法、有意なPSMの電荷状態分布 (1%FDR,配列ベース)
Thunder分離スキームは電荷状態の上限を3+にしただけでなく、1+のプリカーサーに対する有意な一致の数も約82倍に増加させました。これはペプチドの長さの分布からも確認できます。アミノ酸残基長8~13のであるペプチドの同定率は、両手法とも約90%と類似していますが、これは2+が両手法において最も一般的な電荷状態であることを考慮すれば予想外のことではありません。しかし、Thunder分離法で同定された最大長は23であったのに対し、標準的な分離パターンでは64でした。
著者らはNetMHCpan [2] を用いて同定された HLAIps の数を推定しました。我々も、標準および Thunder 分離法によって同定された、長さが 8~13 アミノ酸残基の有意なペプチド配列をすべて抽出し、NetMHCpan-4.1 にかけました。結果は以下の表 2 の通りです。
| Isolation filter | No. predicted HLAIps |
|---|---|
| Standard | 2919 |
| Thunder | 4860 |
Table 2: NetMHCpanによりHLAIpsと予測された、標準およびThunder DDA-PASEF検索結果で同定された長さ8~13のペプチドの数
ご覧の通り、Thunder-DDA-PASEF分離スキームを適用した結果では、予測されるHLAIpsの数が大幅に増加しています。
全体として、データの処理にMascot Distillerを使用し、生成されたピークリストの検索にMascot Server 3.1を用い、機械学習を用いて結果をリファインメント(精緻化)し、さらにMS2PIPからのスペクトル予測情報も利用することで、優れた結果が得られています。
References:
- Gomez-Zepeda D, Arnold-Schild D, Beyrle J, et al. Thunder-DDA-PASEF enables high-coverage immunopeptidomics and is boosted by MS2Rescore with MS2PIP timsTOF fragmentation prediction model. Nat Commun. 2024;15(1):2288. Published 2024 Mar 13. doi:10.1038/s41467-024-46380-y
- Hoof I, Peters B, Sidney J, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61(1):1-13. doi:10.1007/s00251-008-0341-z
Keywords: hla, machine learning, Mascot Distiller, MS2PIP, peak picking, timsTOF