Mascot Serverによる、狭いウィンドウ幅・スペクトル中心的なDIA解析
DIA (Data Independent Acquisition)における分離ウィンドウの幅の広さは、狭いウィンドウ幅と広いウィンドウ幅の2つの戦略に大別されます。また別の観点として、ペプチド中心的な(peptide-centric)手法とスペクトル中心的な (spectrum-centric) 手法の2つのデータ解析戦略があります。通常広いウィンドウ幅のDIA解析はペプチド中心のアプローチである必要がありますが、狭いウィンドウ幅のDIA解析ではどちらの手法も適用可能です。今回のお話の焦点は「狭いウィンドウ幅・スペクトル中心」のDIA解析で、この手法に対してはMascot Serverのキメラスペクトル検索機能を使って実行することが可能です。
Peptide-centric vs. spectrum-centric
ペプチド中心的な検索のコンセプトについては、Tingらの論文[1]でわかりやすく説明されています。ペプチド中心的な検索は、クエリーの実体が実験または予測されたMS/MSスペクトルのライブラリー側のペプチドです。一方スペクトル中心の検索はクエリーの実体がスペクトル側で、これらを計算されたフラグメント質量を観測されたスペクトルと比較します。
(訳者注: 両者の違いについては、紹介論文、特にTable1の比較表やFig.1の解析の流れを表した図がわかりやすいです。また、上記日本語の記述は元のブログ記事英文と少し異なります)
現在のほとんどのDIAソフトウェアはペプチド中心的な手法です。このアプローチは成功していますが、欠点として常にスペクトルライブラリー(in silicoで計算されたものも含む)が必要なことが挙げられます。なおin silicoのライブラリーに対して「ライブラリーフリー」という用語が用いられることがありますが、これは少し誤解を招きやすい表現だと思います。ライブラリーを準備しなければならないということは、(実験ライブラリーや機械学習モデルの選択内容にもよりますが)タンパク質配列の最大数、可変修飾設定、切断ミス許容数などの設定適用に限界があります。また、ライブラリーを生成した装置・測定方法にも注意を払う必要があります。- timsTOFスペクトルライブラリーに対してOrbitrapデータを検索しても、検索がうまくいく可能性は低いでしょう。
対照的に、スペクトル中心の検索はスペクトルライブラリーを必要としないため、ライブラリーや機械学習モデルに関連する限界や仮定に依存しません。 スペクトル中心のマッチングは、各同定が他との比較無しに評価される必要があるため、解釈と視覚化がより簡単です。また、各スペクトルは独立して検索されるため、複数のプロセッサーが存在する場合検索の並列処理が容易です。
狭いウィンドウ幅のDIA解析戦略(分離ウィンドウは一般的に8m/z以下)では、一般的に1つのMS/MSスペクトルデータあたりに含まれるペプチド数が少なくなるため、広いウィンドウ幅(分離ウィンドウ>8m/z)の解析アプローチと比べると複雑なデータにはなりません。 以前のブログ記事(英語版、日本語版)で説明したように、狭いウィンドウ幅のDIAスペクトルは、DDA(Data Dependent Acquisition) の複数ペプチド混入データとあまり変わらないレベルの複雑さに留まると言え、従来のスペクトル中心分析を適用できる可能性があります。
狭いウィンドウ幅のDIAデータセットの解析例
MASCOTのキメラスペクトルの検索に関するヘルプページでもご紹介していますが、Mascot DistillerはThermoFisher Scientific社製質量分析装置で生成された狭いウィンドウ幅のDIAデータファイルを処理することができます。MASCOT関連ソフトウェアによる狭いウィンドウ幅のDIAデータ解析について検証するため、EBI PRIDEレポジトリにて一般公開されているnarrow-window DIAデータセットを取得し、Mascot Distiller 2.8.5とMascot Server 2.8.3を使用した再解析を実施ました。本ブログ記事はその解析結果についてご紹介しています。
選択されたデータセットは前立腺がんに関する研究[2]から得られたものです。前立腺がんと非がん患者のマッチングされた20グループからプロテオミクスデータを収集・解析するため様々なアプローチが行われました。狭いウィンドウ幅[8 m/z]のDIAデータセットです。 先のブログ記事で処理した(DDAにしては)広いウィンドウ幅のDDAデータセットと同じ方法で、(DIAの中では)狭いウィンドウ幅のDIAデータファイルをMascot Distillerを使用して再処理しました。処理の過程ではMascot Daemonを用いて、ピーク抽出と検索実行を自動化しています。ピーク抽出の際に使用した処理オプションは、デフォルト設定としてDistiller側で準備している prof_prof.ThermoXcalibur.opt設定から更に以下の変更を加えたものです:
- スペクトルあたり最大9個のプリカーサー検出を許容し、誤差範囲を+/- 4Da とする。
- 元のスペクトルデータを、1Daあたり400データポイントのデータに変換する(Uncentroiding)。
ピーク抽出処理を行った結果、以下の表1が示すように、大半のピークリストは、プリカーサー候補を1つから3つ検出し、1つのピークリストあたりに検出されたプリカーサーの平均数は約2.5個でした。
| No. of precursors masses | Count |
|---|---|
| 1 | 1293221 |
| 2 | 1150950 |
| 3 | 1978330 |
| 4 | 368796 |
| 5 | 182577 |
| 6 | 77935 |
| 7 | 28403 |
| 8 | 8928 |
| 9 | 3056 |
生成されたピークリストを、Mascot Server 2.8.3に対して検索しました。多価のフラグメントイオンは検索にかける前にDistillerを使ってMH+の値に変換処置をしました。検索に関するパラメーターは論文の内容と揃えています。計算の全プロセスはMascot Daemonを使用して自動化しました。検索完了後、40の生データファイル全てから派生したマスタープロジェクトを生成するため、Mascot Daemonによって作成されたDistillerプロジェクトファイルを統合しました。統合された検索結果に対して、1%のPeptide Spectrum Match False Discovery Rateで同定基準値が調整され、さらに同定タンパク質に対してMascot Distiller[LFQモジュール] を用いて定量解析されました。
1%のPSM FDRを同定基準として定めたところ、データセットから合計で約160万個の有意なPSMs(訳者注:Peptide-Spectrum Matches,「同定スペクトル数」とお考えください。)が検出、4,296個のタンパク質と約20,500個のペプチド配列が同定されました。 結果は以下の表2のようにまとめられます。
| Number of precursors | Significant precursor masses matched per sprectrum | Count |
|---|---|---|
| 1-9 | 1 | 1316967 |
| 2-9 | 2 | 150679 |
| 3-9 | 3 | 5832 |
| 4-9 | 4 | 73 |
| 5-9 | 5 | 1 |
すなわちマッチしなかったピークリストを除くと、1スペクトルあたり約1.1個の有意なプリカーサー質量が同定基準を超えたことがわかります(アサインされなかったスペクトルは除く)。複数プリカーサーがマッチしているケースに対してピーク強度との関係性を確認したところ、やはり最も強い強度ピークのプリカーサーに対して最も多くマッチし、2番目に強いプリカーサーにも妥当な数のマッチが得られていました。しかし3つ以上のデータとなると、スペクトルの複雑さがスコアリングに強い悪影響を及ぼし始めます。下の図1は、2つのプリカーサーの質量と2つの強力なペプチドのマッチを持つキメラスペクトルのマッチングの内訳を示した、わかりやすい例です。
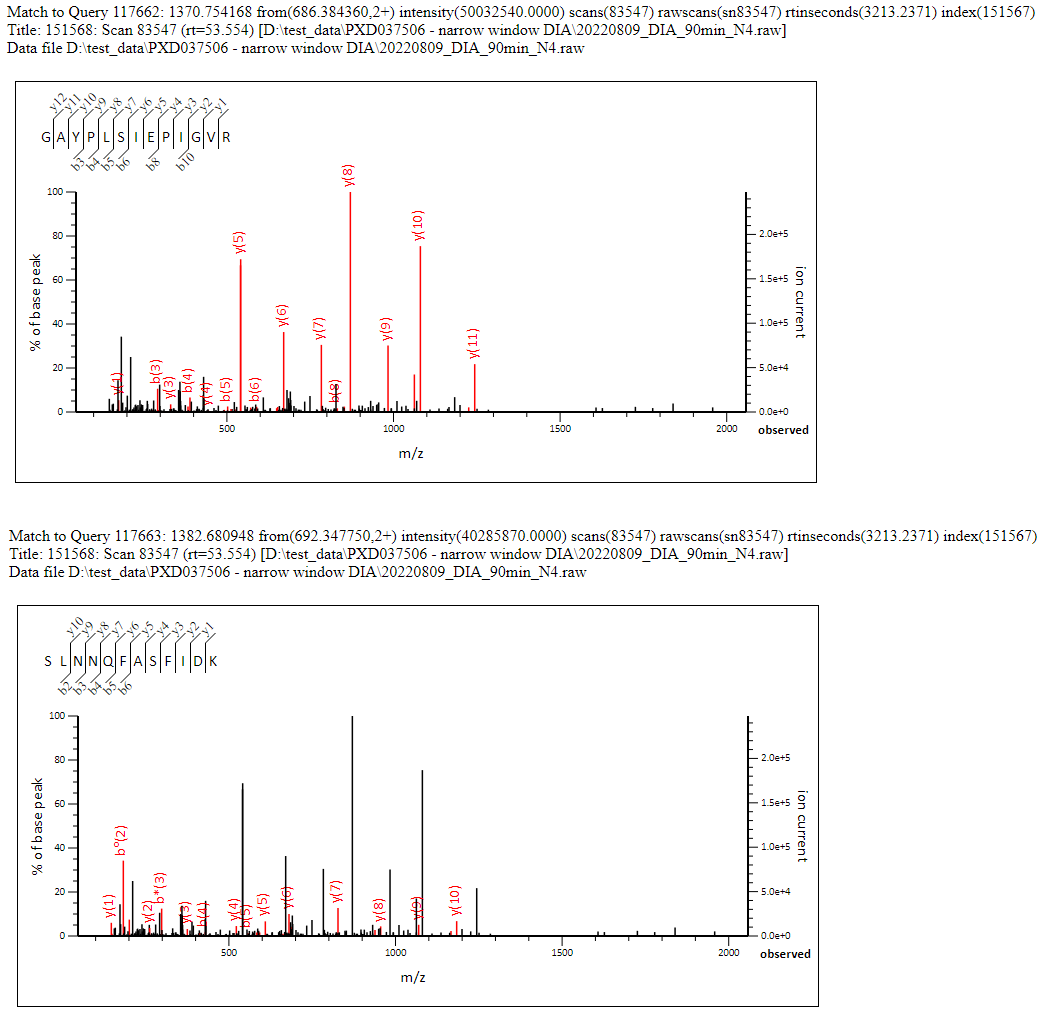
Click to view full size image
図1:キメラスペクトルデータのマッチング例。狭いウィンドウ幅のDIAデータで、2つのプリカーサーがMASCOTにより検出されました。 スペクトル中の強いピークの大部分はこの2つのマッチがカバーしています
定量解析結果
Mascot Distillerを使用し、測定データ間で保持時間の補正を行いながら、データセット全体のラベルフリー定量解析を行いました。下の図2は、解析から得られたペプチドマッチのXICの良い例です。スペクトル中心のアプローチをとっているため、ほとんどのサンプルでXIC領域全体にわたってペプチドがマッチしており、同定の信頼性が高いことがわかります。

Click to view full size image
図2:同一ペプチドのXICの例。データセットに含まれる40サンプルすべてにわたって同じペプチドが検出されています。グラフの灰色の領域はペプチドが同定された箇所を示しています。
原著論文の著者は、前立腺がん患者サンプルにおいて、すべての実験的アプローチで一貫して発現上昇が認められた3つの細胞外小胞(EV)タンパク質を選び出しています。しかし論文の図4だけはその結論とはちょっとあいません。3つのタンパク質がすべての患者サンプルで発現上昇を認める事は難しいです。この3つのEVタンパク質とは、具謡的に以下のものです:
- α1-アンチキモトリプシン(SERPINA3)
- ロイシンリッチα2-糖タンパク質(LRG1)
- セクレトグロビンファミリー 3Aメンバー1(SCGB3A1)
Mascot ServerおよびMascot Distillerを用いた再解析では、前立腺がん患者サンプルの多くでこれら3つのタンパク質において発現が上昇していることが確認できました。Mascot Distillerのレポート機能の1つを使用して作成したボルケーノプロットによる出力例が下図3です。関心のある3つのタンパク質について、plot.ly のライブラリを使用して目立つよう表示しています。
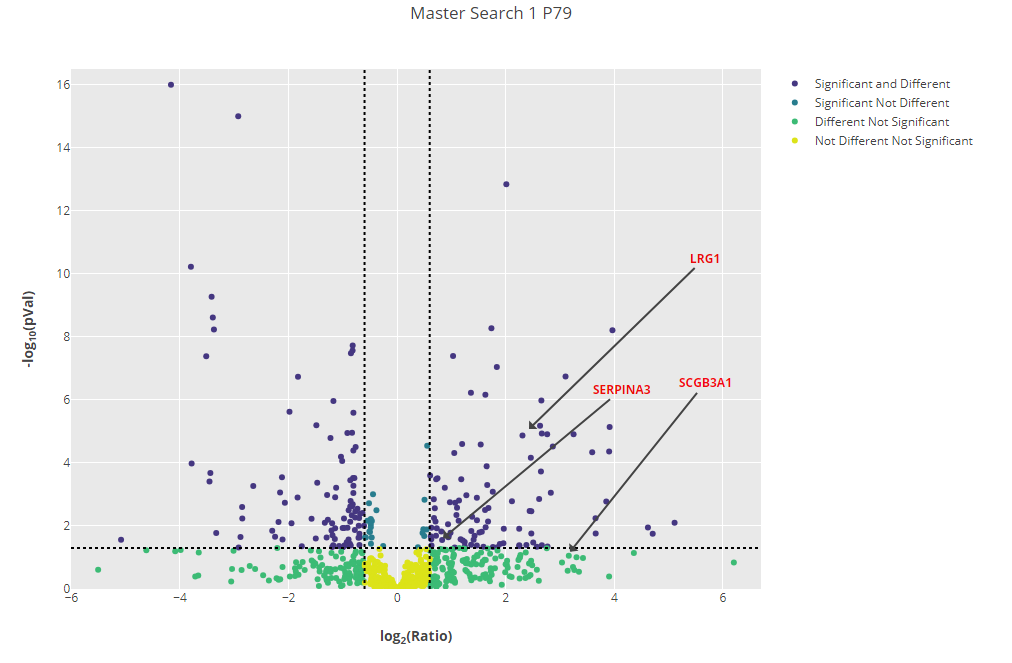
Click to view full size image
図3:元の論文で同定された3つのEVタンパク質を目立つよう表示した、サンプル(P79)の発現量のボルケーノプロット
結果をもう少し詳しく調べるために、定量結果に対して主成分分析(PCA)を行い、レポートを出力しました。主成分分析のレポート作成機能もDistillerに標準搭載されています。分析により20人の患者を第1成分で3つのグループ(それぞれ左、中央、右、と名付けた)に大まかに分けることができました。続いて、どのタンパク質が違いを生み出す原因であるかを調べるため、"ANOVA plus Clustering "レポート作成をDistillerにて実行しました。解析は、分散分析(ANOVA) を実行し、次にグループ間で有意差を示すタンパク質を取り出し、それらのタンパク質について階層的クラスタリング分析を実行する、というものです。ANOVA分析では、多重検定のためのBenjamini-Hochberg補正でp < 0.001の厳しい有意閾値を適用しました。K近傍法を用いて、1つのタンパク質につき2つまでの欠損値をカバーすることができ、それ以上の比率値が欠損している場合は当該タンパク質を解析から除外しました。結果として得られた階層的クラスタリングのレポートが下図4です。 ご覧の通りANOVAによって、3つの異なるグループは同定されたタンパク質の発現量により非常に明確に分かれました。 例えばROBO4[3]など。そしてこれらのタンパク質の多くは、前立腺がんや他のがんに関連することが知られているものでした。
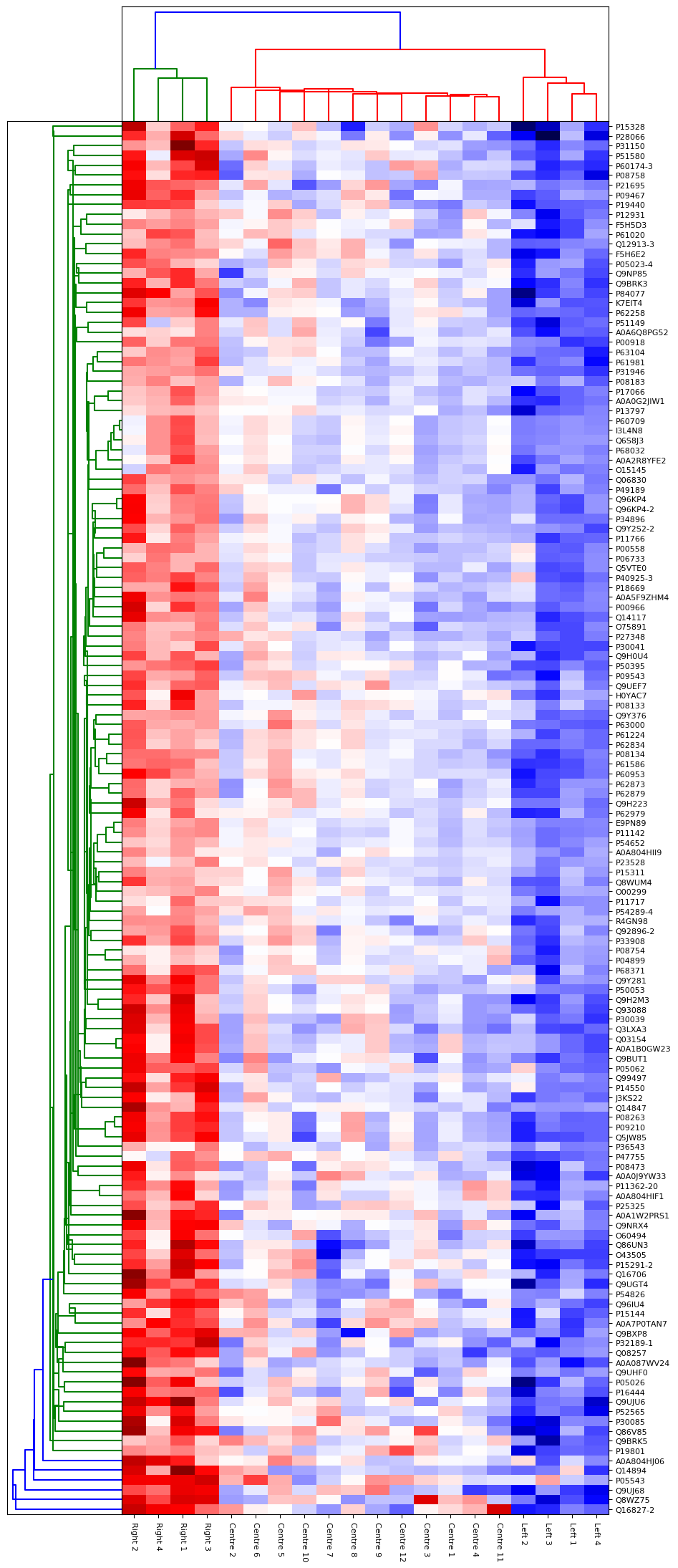
Click to view full size image
図4:PCAによって同定された患者の3つのグループ間でANOVAによって有意に異なると同定されたタンパク質の階層的クラスタリング。
Percolator
感度を上げる(この場合、同定ペプチド数を増やす)方法の1つとして、Percolatorを使うことが挙げられます。Percolatorは、スペクトルや同定されたペプチドから抽出したそれらの特徴を使用して、マッチング内容を再スコアリングします。 Mascotの現在のリリースでは、単一の結果ファイルに対してのみPercolatorのスコアリングを適用することができます。しかしMASCOT側で結合した結果に対してPercolator を適用する事ができません。Distillerで定量解析を行う際にも同様の制限により、Percolatorが適用できません。この件についてはMascot Serverの次のリリースで対処する方向で検討しています。現状複数ファイルの場合に適用が出来ない中、Percolator適用による効果の検討をするため、プロジェクトのN4サンプル(5つの有意なPSMを持つスペクトルを持つファイル)から結果を取り出し、Percolator適用なしとありの結果を比較しました。その結果を以下の表3にまとめています。
| No. matched precursors | N4 significant PSMs | Percolator | |
|---|---|---|---|
| 1 | 34436 | 47967 | |
| 2 | 3759 | 8917 | |
| 3 | 147 | 1089 | |
| 4 | 2 | 58 | |
| 5 | 1 | 2 | |
| No. peaklists with 1 or more significant precursor matches | 38345 | 47967 | |
| Total no. significant PSMs | 42408 | 59244 | |
| Mean no. sig. PSMs / matched spectrum | 1.11 | 1.24 |
ご覧のように、パーコレーターのリスコアリングを使用することで、明らかに有意なPSMの数が大幅に増加します。42,408から59,244と、16,836、40%程度の増加です。より多くのキメラマッチが得られている今回のようなケースでは、Percolatorのリスコアリングにより、複雑なスペクトルのマッチングにおける感度を向上させる事が期待できます。
結論
今回行ったスペクトル中心の解析は概ね成功し、元の論文で報告されたものと遜色のない検索結果と定量結果が得られました。スペクトル中心のアプローチをとることで、MS/MSデータに対して標準的なデータベース検索を実行することができ、実測または予測されたスペクトルライブラリーを準備しなければならない必要性を回避することができます。このアプローチは、従来問題点として指摘されている修飾に関する検索を行う上で明らかな利点があります。実験ライブラリーを使ったアプローチではライブラリーの中に必ず該当する修飾がついたペプチドをデータ内に含んでいる必要があります。また予測ライブラリーデータを使う場合でもモデルに使用するトレーニングデータに目的の修飾に対応しておく必要があります。プリカーサースキャンを使ってペプチドを同定・定量することで、ペプチドの予測保持時間などを計算する必要がありません。
もちろん、このアプローチにも欠点はあります。 MascotとDistillerはどちらも主にDDAワークフロー用に設計されているため、今回報告したようにデータをまとめる機能が不足しています。またこのようなデータセットを扱うには、大容量のメモリを搭載した非常に高速なコンピューターが必要になります。さらに、有意なマッチングとして認識できるスペクトルの複雑さにも限界があります。Mascotのキメラスペクトラムマッチングを、より複雑で広いウィンドウのDIAデータを扱うためにさらなる改良をする必要があるでしょう。また目下の課題として、マージされた結果に対してPercolatorによる再スコアリングを適用できるようにする必要があります。そうする事で明らかにマッチングと定量結果の感度を向上させる事ができます。
今後のMascot ServerおよびMascot Distillerのリリースを重ね、このような制限の多くを改善すると共に、多くの装置分析装置メーカーのRawファイル読み込みや、ワークフローからのDIAデータへの対応を強化する予定です。
参考文献
- [1] Ting Y S et al. Molecular and Cellular Proteomics 2015, 14(9), 2301-2307. doi:10.1074/mcp.O114.047035
- [2] Zhang H et al. Molecules 2022, 27(23), 8155. doi:10.3390/molecules27238155
- [3] Pircher A et al. Int J Med Sci 2019, 16(1), 115-124. doi:10.7150/ijms.28735
Keywords: chimeric spectra, DIA, label-free, LFQ, Mascot Distiller, Thermo